
Application Note
Validate CRISPR-Edited Cells using Imaging and Western Blot Detection on a Microplate Reader
- Measure transfection efficiency by accurately identifying GFP-positive cells vs non-transfected cells
- Validate CRISPR-mediated gene knockdown using western blot analysis on protein bands
- Capture high-quality western blot images
Introduction
Genome editing has been widely used to study gene expression and protein function, but many of these methods are labor intensive and inaccurate1 . The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 system has become a very popular tool for editing genes due to its high accuracy and ease of use2. It originated as part of the innate immune system of bacteria and archaea and was used to protect against foreign DNA3,4. Short sequences of known foreign DNA are expressed as CRISPR targeting RNA (crRNA), which guide the Cas9 enzyme to cleave any foreign DNA containing a similar sequence. By harnessing this system, researchers can study the effects of gene silencing and transcription regulation, and they can potentially alleviate genetic disorders in diseased cells5.
A guide RNA (gRNA) similar to a crRNA is designed to target a region in the gene, and the Cas9 enzyme can create doublestrand breaks in this specific region of the host cell’s genome (Figure 1). After a double-strand break is made, the cell will undergo one of two repair pathways: the nonhomologous end joining (NHEJ) pathway or the homology-directed recombination (HDR) pathway. The NHEJ pathway is commonly used to disrupt genes via base insertions or deletions (indels), while the HDR pathway can be used to knock in a reporter gene or an edited sequence by exchanging sequences between two similar or identical molecules of DNA6.
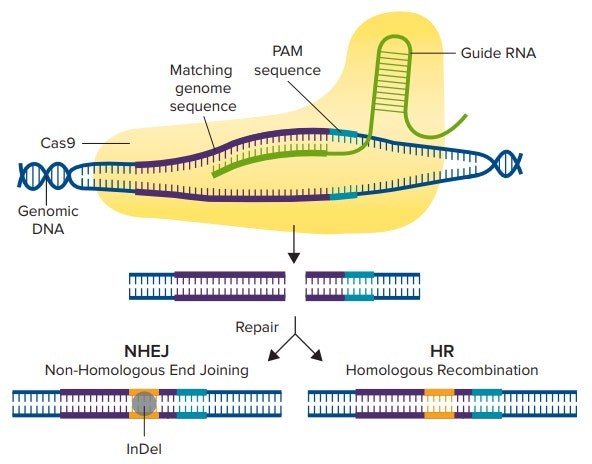
Figure 1. CRISPR/Cas9 Mechanism. The Cas9 enzyme is activated by first binding to a guide RNA, then binding to the matching genomic sequence that immediately precedes 3-nucleotide PAM sequence. The Cas9 enzyme then creates a double-strand break, and either the NHEJ or the HDR pathway is used to repair the DNA, resulting in an edited gene sequence.
Validation of gene editing is necessary to determine if the introduced mutation knocked down the gene of interest. Usually, genomic PCR and genetic sequencing tests are performed to confirm successful deletions or insertions, however, measuring protein expression is a more definitive measure of gene knockdown, avoiding signal from in-frame deletions/mutations7 .
Here, we demonstrate how the SpectraMax i3x Multi-Mode Microplate Reader and ScanLater Western Blot Detection System can be used to validate CRISPR/Cas9 genome editing. We used Origene’s ATG5 human gene knockout kit via CRISPR kit to knock down Autophagy Related 5 (ATG5) protein expression in HEK293 cells and knock in green fluorescent protein (GFP) and puromycin resistance genes. Knockdown was verified with ScanLater western blot analysis.
Materials
- ATG5 Human CRISPR Knockout kit via CRISPR (Origene cat. #KN210563)
- FUGENE HD Transfection Reagent (Promega cat. #E2311)
- Opti-MEM Reduced Serum Media (ThermoFisher cat. #31985062)
- Rabbit Polyclonal Antibody against ATG5 (Origene cat. #TA301481)
- ATG5 HEK293T cell transient overexpression lysate (Origene cat. #LC401532)
- pCas-Guide-EF1a-GFP Plasmid (Origene cat. #GE100018)
- HEK293 cells (ATCC cat. #CRL-1573)
- SpectraMax® i3x Multi-Mode Microplate Reader (Molecular Devices cat. #i3x)
- ScanLater™ Western Blot Detection System (Molecular Devices cat. #0200-7027)
- SpectraMax® MiniMax™ 300 Imaging Cytometer (Molecular Devices cat. #5024062)
- ScanLater: Eu-Streptavidin Evaluation Kit (Molecular Devices cat. #R8200)
- ScanLater: Anti-Mouse Evaluation Kit (Molecular Devices cat. #R8201)
- Pierce BCA Protein Assay Kit (Thermofisher cat. #23225)
- Puromycin (InvivoGen cat. #ant-pr)
- TGX 12% SDS-PAGE Gel (Bio-Rad cat. #1620174)
- TransBlot Turbo Mini-size LF PVDF membranes (Bio-Rad cat. #1620174)
- Trans-Blot® Turbo™ system (Bio-Rad cat. #1704150)
Methods
Cell transfection
HEK293 cells were seeded in Costar tissue culture-treated 6-well plates at 300,000 cells per well. Cells were transfected with (1) a guide vector (AAGATGTGCTTCGAGATGTG) and (2) a donor vector containing sequences for puromycin resistance and GFP. In parallel, three wells of cells were transfected with the pCas-Guide-EF1a-GFP vector that transiently expresses GFP as a transfection control. 6 µL of FUGENE HD transfection reagent and 2 µg of total DNA (3:1 ratio) were used to transfect cells. Afterwards, cells were incubated at 37°C overnight. Transfected cells expressed GFP and were identified and counted in the green fluorescence channel of the MiniMax cytometer. Total cell count was calculated using StainFree analysis, and transfection efficiency was measured by dividing the number of GFP-positive cells by the total number of cells using SoftMax Pro Software.
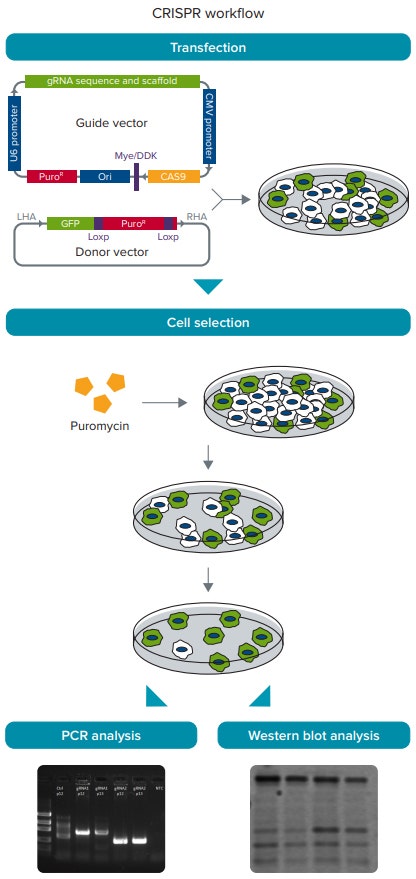
Figure 2. CRISPR/Cas9 experimental workflow. HEK293 cells were transfected with the guide and donor vectors. Cells are then selected with 1.0 µg/mL puromycin to enrich for cells with gene insertion. DNA and protein are collected from the selected cells, and genomic PCR and western blotting are performed to verify gene insertion and knockdown, respectively.
Cell selection
Transfected cells were cultured in medium containing 1.0 µg/mL puromycin to select for cells containing an inserted puromycin-resistance gene. Afterwards, cells were lysed, and genomic DNA and total protein were collected. Genomic DNA was sent to GENEWIZ for PCR analysis to verify correct gene insertion. Protein concentrations were quantitated using the Pierce BCA assay read on the SpectraMax i3x reader, and results were analyzed using a preconfigured protocol in SoftMax Pro Software.
Western blot
5 µg and 10 µg of total protein from both CRISPR-edited cells and unedited cells were prepared by diluting cell lysates in Laemmli sample buffer and boiling at 100°C for 10 minutes. Samples were then loaded onto a 4-20% pre-cast gel, and SDSPAGE was performed. Proteins were then transferred to a PVDF membrane using the Trans-Blot Turbo system’s semi-dry method.
The membrane was incubated in ScanLater Blocking Buffer at room temperature for one hour. The loading control, ladder, and ATG5 region lanes of the membrane were then cut into separate pieces and incubated in their respective primary antibodies (see Table 1) at 4°C overnight. Following three washes in ScanLater Wash Buffer, the blot pieces were then incubated with ScanLater Eu-labeled secondary antibodies at room temperature for one hour. The antibodies and their dilutions are shown in Table 1.
Table 1. Antibody dilutions used for western blot.
Following the secondary antibody incubation, the membrane was washed three times in ScanLater Wash Buffer, dried, and reassembled before reading with the ScanLater Western Blot Detection System.
Data analysis
Data were exported from SoftMax Pro Software into ImageJ software, and band densities were measured to quantitate relative ATG5 protein expression. Vinculin, a 117-kDa cytoskeletal protein associated with cell-cell and cell-matrix junctions, was used as a loading control to normalize for sample loading variation.
Results
The MiniMax cytometer captured high quality images of both transfected and non-transfected cells, and SoftMax Pro Software was able to identify and quantitate the number of GFP-positive cells and total number of cells. A 13% transfection efficiency was calculated (Figure 3).
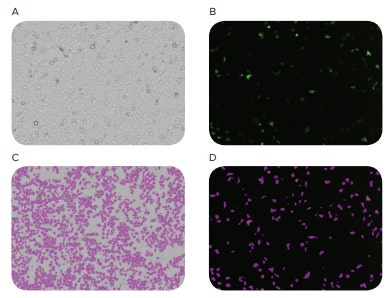
Figure 3. Images of cellular transfection. HEK293 cells were transfected with a pCas-Guide-EF1a-GFP vector in parallel with CRISPR/Cas9-transfected cells to calculate transfection efficiency. The MiniMax cytometer was used to image cells in the bright-field channel (A) and the green fluorescence channel (B). SoftMax Pro Software identified and counted cells in both channels (C and D), and a 13% transfection efficiency was calculated by dividing GFP-positive cells by total cell count.
PCR analysis of genomic DNA samples determined that the correct-sized insertion had been made in the ATG5 region of the HEK293 cell genome (data not shown).
In the western blot image, visible decrease in ATG5 protein band intensity was seen between the CRISPR-edited cells and the unedited cells (negative control) (Figure 4). Using ImageJ software to calculate protein band density, CRISPR-edited cells had roughly 60% knockdown in ATG5 protein expression compared to the unedited cells (Figure 5).
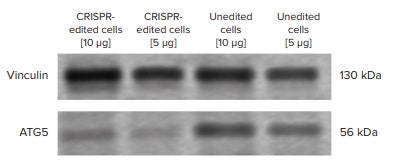
Figure 4. ATG5 western blot experiment. ATG5 protein expression was visibly lower in CRISPRedited cells compared to unedited cells. 5 µg and 10 µg of sample were loaded onto the SDS-PAGE gel to account for over/under sample loading. Vinculin was used as a loading control to normalize for variable sample loading. The western blot image was exported to ImageJ software to measure band density.
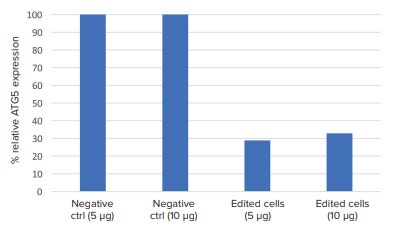
Figure 5. Relative ATG5 protein expression. The western blot image from Figure 4 was exported to ImageJ software, and band density was analyzed. ATG5 protein band density was normalized to their respective loading controls. CRISPR-edited cells had consistently lower ATG5 protein expression compared to non-edited cells.
Conclusion
CRISPR gene editing technology requires careful monitoring of the entire process to ensure accurate results. The SpectraMax i3x Multi-Mode Microplate Reader provides a complete solution for analyzing results of a CRISPR-editing experiment from initial transfection to confirmation of protein knockdown. With the MiniMax cytometer, researchers can assess transfection efficiency by comparing total unlabeled cell counts to counts of fluorescenceexpressing tranfected cells. The ScanLater Western Blot Detection System enables sensitive detection and quantitative analysis of proteins of interest in control and CRISPR-edited cells.
References
- Lombardo, A et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature Biotechnology 25.11 (2007): 1298.
- Barrangou, R. Cas9 targeting and the CRISPR revolution. Science 344.6185 (2014): 707-708.
- Ishino, Y et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology 169.12 (1987): 5429-5433.
- Barrangou, R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315.5819 (2007): 1709-1712.
- Barrangou, R et al. Applications of CRISPR technologies in research and beyond. Nature Biotechnology 34.9 (2016): 933-941.
- Liang, F et al. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proceedings of the National Academy of Sciences 95.9 (1998): 5172-5177.
- Estep, JA et al. Immunoblot screening of CRISPR/Cas9-mediated gene knockouts without selection. BMC molecular biology 17.1 (2016): 9.
简介
基因组编辑已被广泛应用于基因表达和蛋 白质功能的研究,但这些方法中有许多是 复杂的和不准确的[1]。CRISPR ( 成簇的规 律间隔的短回文重复序列 ) / Cas9 系统由于 其较高的准确性和易操作性,已经成为一 种非常流行的基因编辑工具[2]。它起源于 细菌和古细菌先天免疫系统的一部分,被 用来抵御外来的 DNA[3,4]。已知的外源 DNA 短序列被表达为 CRISPR 靶向 RNA (crRNA),引导 Cas9 酶切割任何含有类似 序列的外源 DNA。通过利用这一系统,研 究人员可以研究基因沉默和转录调控的效 果,并有可能减轻患病细胞的遗传疾病[5]。
一个与 crRNA 相似的向导 RNA (gRNA) 被 设计针对基因的某一段区域,Cas9 酶可以 在宿主细胞基因组的指定区间制造双链断 裂 ( 图 1 )。双链断裂后,细胞会经历两种 修复途径;非同源末端连接 (NHEJ) 途径 和同源定向重组 (HDR) 途径。NHEJ 途径 是通常被用于破坏基因,通过插入或删除 ( 位点 ),而 HDR 途径可以被用于敲除报 告基因或通过在两个相似或相同的序列之 间交换序列而编辑的序列[6]。
图 1 CRISPER/Cas9 机制。 Cas9 酶被首先结合的向导 RNA 激活,然后结合到包含 3-核苷酸 PAM 序 列的匹配的基因上。Cas9 酶随后创建了一个双链断裂,通过 NHEJ 和 HDR 途径修复基因,生成一 个编辑后的基因序列。
基因编辑的确认是必要的,为了确定引入 的变化是否破坏了感兴趣的基因。通常会 用染色体 PCR 和基因的测序来确认是否成 功的删除或插入,然而检测蛋白的表达是 基因敲除更好的检测方式,避免结构内删 除 / 突变[7]。
这里我们展示了如何利用 SpectraMax i3x 多功能微孔板读板机和 ScanLater 免疫印迹 检测卡盒鉴定 CRISPR/Cas9 基因编辑。我 们使用 Origene 公司的 ATG5 人源基因敲 除试剂盒通过 CRISPR 试剂盒敲除 HEK293 细胞中自噬相关的 ATG5 蛋白表达并插入 绿色荧光蛋白 (GFP) 和抗嘌呤霉素基因。 敲除是使用 ScanLater 免疫印迹鉴定。
材料
- ATG5 Human CRISPR Knockout kit via CRISPR (Origene cat. #KN210563)
- FUGENE HD Transfection Reagent (Promega cat. #E2311)
- Opti-MEM Reduced Serum Media (ThermoFisher cat. #31985062)
- Rabbit Polyclonal Antibody against ATG5 (Origene cat. #TA301481)
- ATG5 HEK293T cell transient overexpression lysate (Origene cat. #LC401532)
- pCas-Guide-EF1a-GFP Plasmid (Origene cat. #GE100018)
- HEK293 细胞(ATCC cat. #CRL-1573)
- SpectraMax® i3x 多功能微孔板读板机 (Molecular Devices cat. #i3x)
- ScanLater™ Western Blot 检测系统 (Molecular Devices cat. #0200-7027)
- SpectraMax® MiniMax™ 300 细胞成像 系统 (Molecular Devices cat. #5024062)
- ScanLater: Eu-Streptavidin Evaluation Kit (Molecular Devices cat. #R8200)
- ScanLater: Anti-Mouse Evaluation Kit (Molecular Devices cat. #R8201)
- Pierce BCA Protein Assay Kit (Thermofisher cat. #23225)
- Puromycin (InvivoGen cat. #ant-pr)
- TGX 12% SDS-PAGE Gel (Bio-Rad cat. #1620174)
- TransBlot Turbo Mini-size LF PVDF membranes (Bio-Rad cat. #1620174)
- Trans-Blot® Turbo™ system (Bio-Rad cat. #1704150)
方法
细胞转染
将 HEK293 细胞以每孔 300,000 个细胞的 密度种在 Costar 6 孔组织培养板中。细胞 (1) 被向导载体 (AAGATGTGCTTCGAGATGTG) 和包含嘌呤霉素抗性和 GFP 的供体 载 体 转 染 。 平 行 的 , 三 个 孔 的 细 胞 被 pCas-Guide-EF1a-GFP 载体转染,该载体 瞬时表达 GFP 作为转染控制。6 µL FUGENE HD 转染试剂和 2 µg 总 DNA (3:1) 用于转染 细胞。然后将细胞放置在 37℃ 孵育过 夜。表达 GFP 的被转染的细胞可以通过 MiniMax cytometer 的绿色荧光通道鉴定 和计算。细胞的总数可以通过免染分析被 计算,转染效率可以通过 SoftMax Pro 软 件计算 GFP 阳性的细胞数与总细胞数的比 值。
图 2 CRISPER/Cas9 实验流程。HEK293 细胞被向导和供体载体转染。 使用 1.0 µg/mL 嘌呤霉素 选择富集基因插入的细胞。在被选择的细胞里收集细胞的 DNA 和蛋白,基因组 PCR 和免疫印迹分 别用于鉴别基因的插入和敲除。
细胞挑选
转染的细胞被培养在含有 1.0 µg/mL 嘌呤 霉素的培养基当中以挑选插入嘌呤霉素抗 性基因的细胞。然后,裂解细胞,收集基 因组 DNA 和总蛋白。基因组 DNA 被送去 GENEWIZ 做 PCR 分析鉴定基因插入的正 确性,使用 Pierce BCA 实验在 SpectraMax i3x 读板机上定量蛋白浓度,并使用 SoftMax Pro 软件中的一个预置模板分析 结果。
免疫印迹
在 Laemmli 样品缓冲液中稀释细胞裂解 物,在 100℃ 下煮沸 10 分钟,从 CRISPER 编辑的细胞和未被编辑的细胞中收集 5 µg 和 10 µg 的总蛋白。上样品至 4-20% 的预制 凝胶上,做 SDS-PAGE。然后使用 TransBlot Turbo 的半干法,将蛋白转移至 PVDF 膜上。
将膜放在 ScanLater 封闭缓冲液中室温孵 育 1 小时。膜上的对照,参比和 ATG5 条带 被剪切成独立片放在各自的一抗 ( 见表 1 ) 中 4℃ 孵育过夜。用 ScanLater 清洗缓冲 液洗三遍以后,再放入 ScanLater Eu 标记 的二抗溶液中室温孵育 1 小时。对应的抗 体和稀释见表 1。
Table 1. Antibody dilutions used for western blot.
二抗孵育过后,用 ScanLater 清洗缓冲液 洗 3 遍,干燥,重新拼接后用 ScanLater 免疫印迹检测系统读板。
数据分析
数据从 SoftMax Pro 软件中导出至 Image J 软件,通过条带亮度定量 ATG5 蛋白的相 对表达。Vinculin 是一种 117-kDa 的细胞 骨架蛋白,与细胞-细胞和细胞-基质连接 有关。该蛋白作为上样对照使样品上样差 异标准化。
结果
MiniMax cytometer 获取转染和非转染细 胞的高质量的图片,SoftMax Pro 软件可 以定量 GFP 阳性的细胞数量和总的细胞数 量。计算得出 13% 的转染效率 ( 图 3 )。
图 3 细胞转染图像。 HEK293 细胞被 pCas-Guide-EF1a-GFP 载体和 CRISPR/Cas9 平行转染来计算转 染效率。MiniMax cytometer 用于在明场通道 (A) 和绿色荧光通道 (B) 获取细胞的图像。SoftMax Pro 软件在两通道内鉴定和计算 ( C 和 D ),通过计算 GFP 阳性细胞与总细胞数的比值算出转染效率为 13%。
PCR 分析基因组 DNA 样品表明,在 HEK293 细胞基因组的 ATG5 区域进行了 正确大小的插入 ( 数据未显示 )。
在免疫印迹图像中,CRISPER 编辑的细胞 与非编辑的细胞 ( 阴性对照 ) 样品在 ATG5 区域显示出明显较弱的条带亮度 ( 图 4 )。 使用 Image J 软件计算蛋白条带的亮度, 与非编辑的细胞相比,CRISPER 编辑的细 胞的 ATG5 蛋白表达下调 60% ( 图 5 )。
图 4 ATG5 免疫印迹实验。 ATG5 蛋白在 CRISPER 编辑细胞内的表达表达明显低于非编辑细胞。10 µg 和 5 µg 样本被上样到 SDS-PAGE 胶上计算过量 / 低量样本。Vinculin 作为对照上样使不同的样本 上样量标准化。免疫印迹图片被导出到 Image J 软件计算条带的亮度。
图 5 ATG5 蛋白相对表达。 图 4 中的免疫印迹图片被导出到 Image J 软件中,分析条带亮度。ATG5 蛋白条带的亮度标准化到他们各自的上样对照。CRISPER 编辑的细胞中的 ATG5 表达一致低于非编辑 细胞。
总结
CRISPER 基因编辑技术需要对整个过程进 行细致的监控以确保实验结果。SpectraMax i3x 多功能微孔板读板机为结果的分析 提供了完整的解决方案。从最初的转染到 蛋白敲除的确认。使用 MiniMax cytometer,研究人员可以通过比对未标记的细胞 总数和表达荧光的转染细胞数评估转染效 率。ScanLater 免疫印迹检测系统可以对对 照蛋白和 CRISPER 编辑的细胞进行敏感检 测和定量分析。
参考文献
- Lombardo, A et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature Biotechnology 25.11 (2007): 1298.
- Barrangou, R. Cas9 targeting and the CRISPR revolution. Science 344.6185 (2014): 707-708.
- Ishino, Y et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology 169.12 (1987): 5429-5433.
- Barrangou, R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315.5819 (2007): 1709-1712.
- Barrangou, R et al. Applications of CRISPR technologies in research and beyond. Nature Biotechnology 34.9 (2016): 933-941.
- Liang, F et al. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proceedings of the National Academy of Sciences 95.9 (1998): 5172-5177.
- Estep, JA et al. Immunoblot screening of CRISPR/Cas9-mediated gene knockouts without selection. BMC molecular biology 17.1 (2016): 9.